De Novo Design Workflow
Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement

Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement
Schrödinger’s De Novo Design Workflow is a fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement. Starting from a hit molecule or lead series, the technology identifies synthetically tractable molecules that meet key project criteria by combining multiple compound enumeration strategies with an advanced filtering cascade (AutoDesigner) and rigorous potency scoring with free energy calculations (Active Learning FEP+).
Through built-in reaction-based enumeration combined with advanced filtering to rule out undesired and unrealistic chemistry
By leveraging accurate potency predictions combined with active learning
By efficiently evaluating up to billions of project-relevant virtual molecules
The De Novo Design Workflow offers a cloud-deployable solution with the flexibility to customize settings and property space for the unique needs of your program.
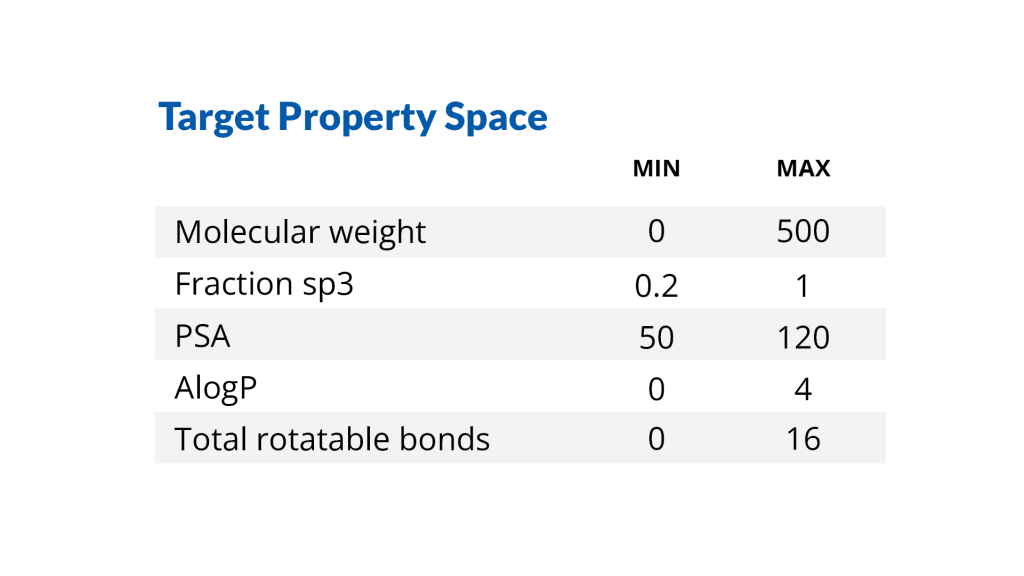
Define the starting molecule, the portion of the molecule to explore, the desired physicochemical property space, and additional project-specific filters within LiveDesign, a web-based enterprise molecular design and collaboration platform.
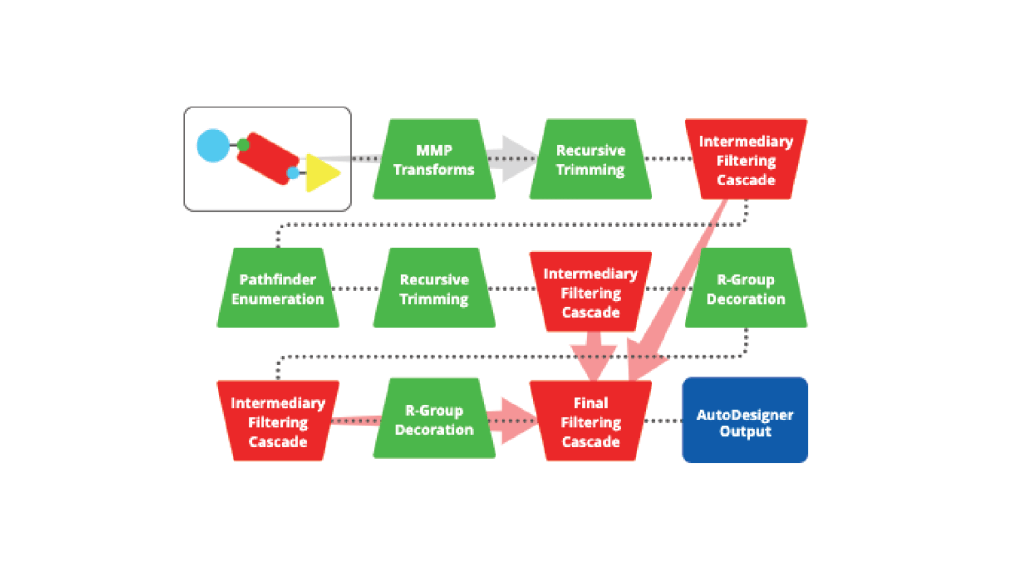
Automatically carry out successive rounds of compound generation and filtering within desired chemical space using cloud-native, multi-stage enumeration strategies combined with an advanced filtering cascade based on physical properties, amenability to FEP+, IP, and docking.
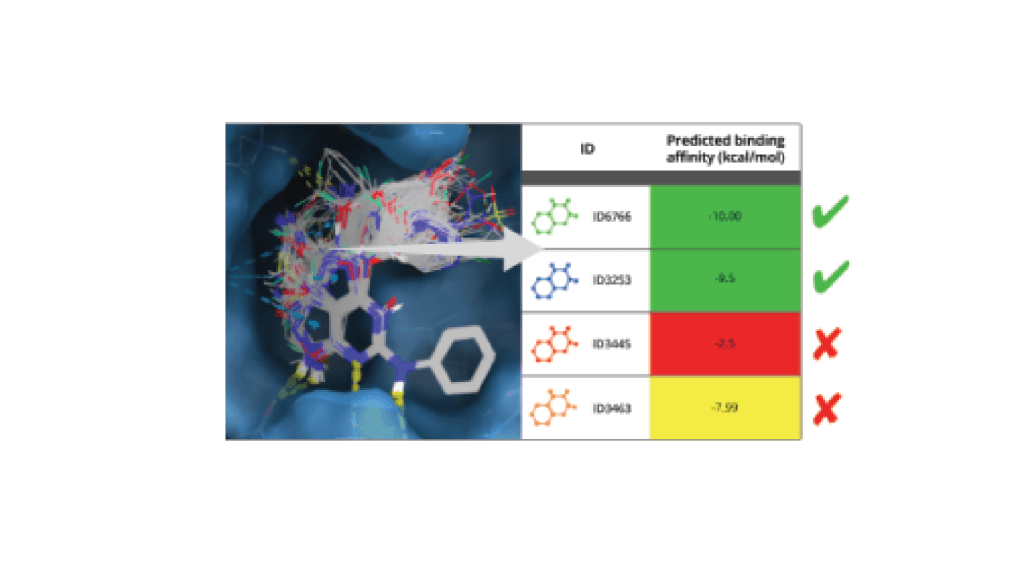
Leverage a well-validated, automated workflow which trains a machine learning model on project-specific FEP+ data to allow processing of up to millions of compounds with highly accurate FEP+ calculations efficiently.
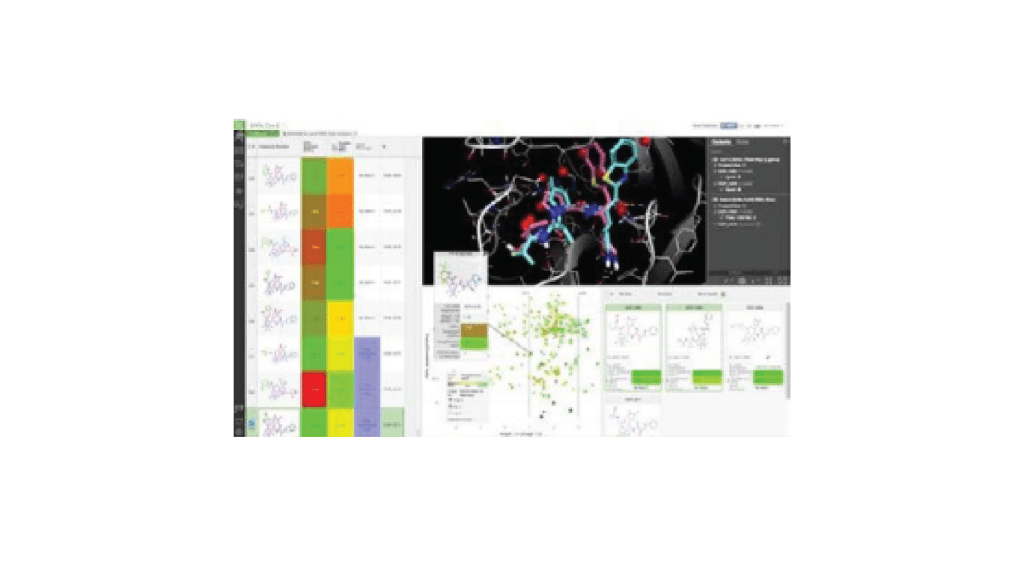
Review the top scoring compounds and use the FEP+-trained machine learning model in LiveDesign — allowing evaluation, interactive optimization, and prioritization by the project team.
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.

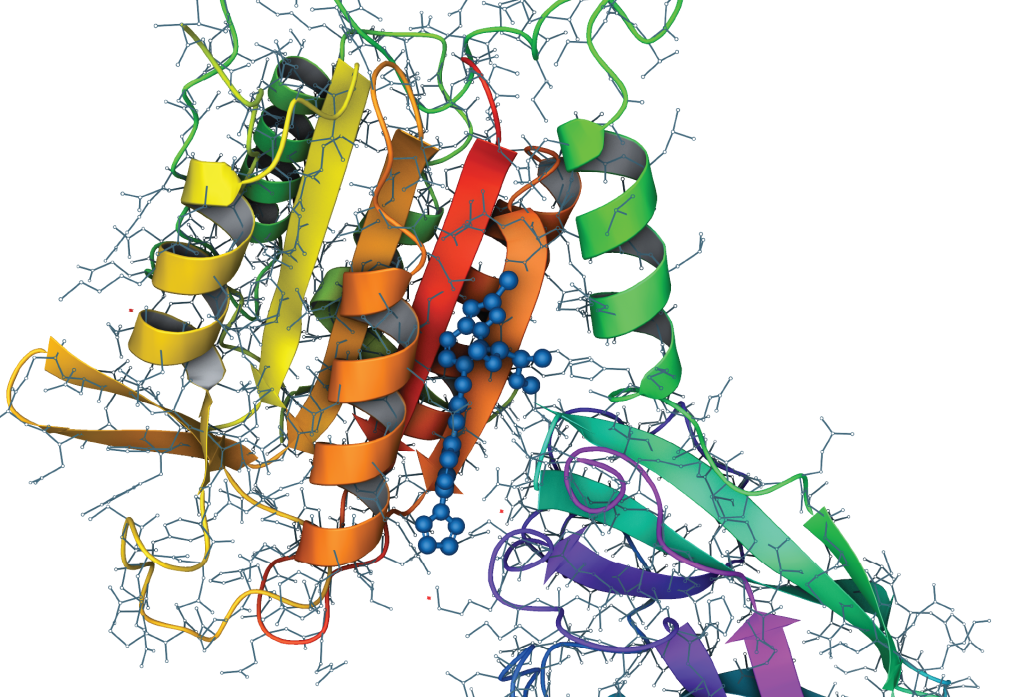
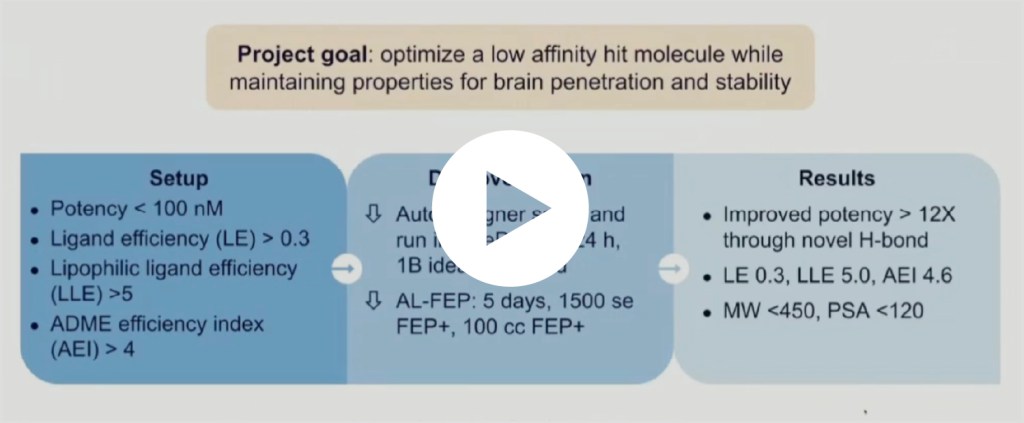
In a recent MALT1 drug discovery program led by Schrödinger’s Therapeutics Group, the De Novo Design Workflow amplified the team’s design efforts.
view case studyLevel up your skill set with hands-on, online molecular modeling courses. These self-paced courses cover a range of scientific topics and include access to Schrödinger software and support.
Learn how to deploy the technology and best practices of Schrödinger software for your project success. Find training resources, tutorials, quick start guides, videos, and more.