
Peptide Discovery

Explore the benefits of computationally-guided peptide design

Enumerate and ideate beyond initial natural peptides on a large scale
Explore mutation to standard and non-standard amino acids, custom backbone modifications, and a wide variety of cyclizations to create novel natural peptides or peptidomimetics with desired properties.
Explore conformational landscapes
Predict 3D structure, calculate relative binding free energies of structurally diverse conformations, and select the most plausible binding mode.
Predict key properties of peptides
Guide peptide discovery by facilitating the design and optimization of peptide structures with desired properties and the associated physicochemical characteristics, such as membrane permeability and solubility.
Understand peptide-target interactions
Employ specialized docking tools, molecular dynamics, and advanced free energy perturbation calculations to predict binding affinity, selectivity, thermostability, and pH-dependencies at accuracies approaching experimental level.
Design high-quality biologics with Schrödinger’s cutting-edge software
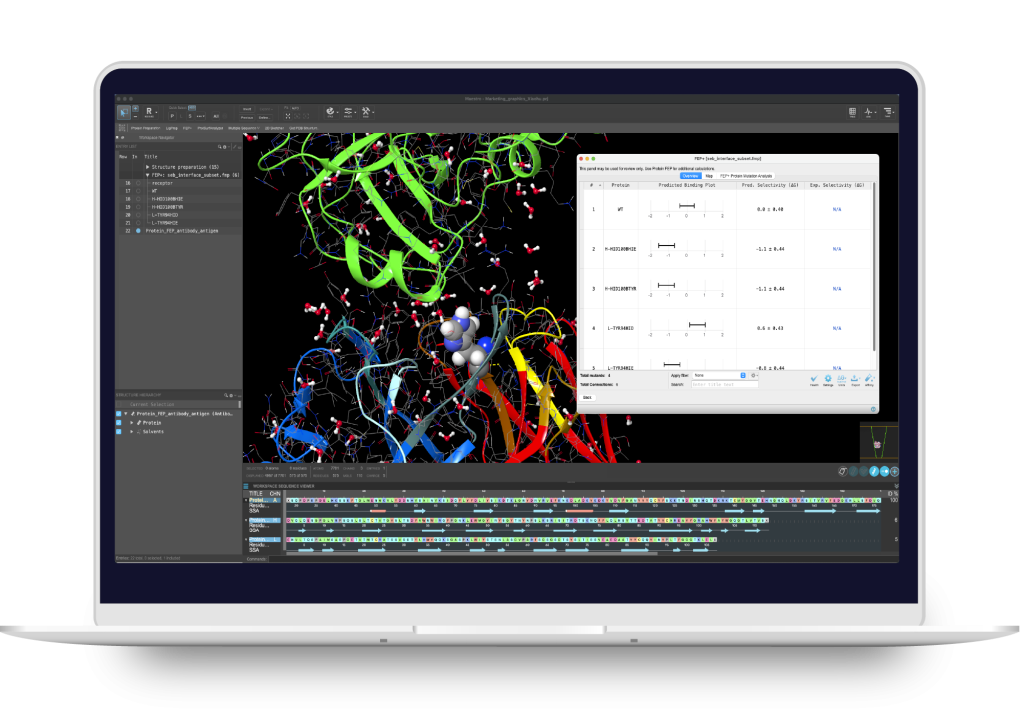
BioLuminate
Modeling environment for biologics discovery
LiveDesign
Collaborative digital biologics design and discovery lab
Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Potency- and Selectivity-Enhancing Mutations of Conotoxins for Nicotinic Acetylcholine Receptors Can Be Predicted Using Accurate Free-Energy Calculations
Dana Katz, et al. Mar. Drugs. 2021, 19(7), 367.
Evaluation of Free Energy Calculations for the Prioritization of Macrocycle Synthesis
Janet L. Paulsen, et al. J. Chem. Inf. Model. 2020, 60, 7, 3489–3498.
Potency-Enhancing Mutations of Gating Modifier Toxins for the Voltage-Gated Sodium Channel NaV1.7 Can Be Predicted Using Accurate Free-Energy Calculations
Dana Katz, et al. Toxins 2021, 13(3), 193.
Key Products
Learn more about the key computational technologies available to progress your research projects.
Software and services to meet your organizational needs
Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Services
Leverage Schrödinger’s computational expertise and technology at scale to advance your projects through key stages in the drug discovery process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.