
Hit-to-Lead & Lead Optimization
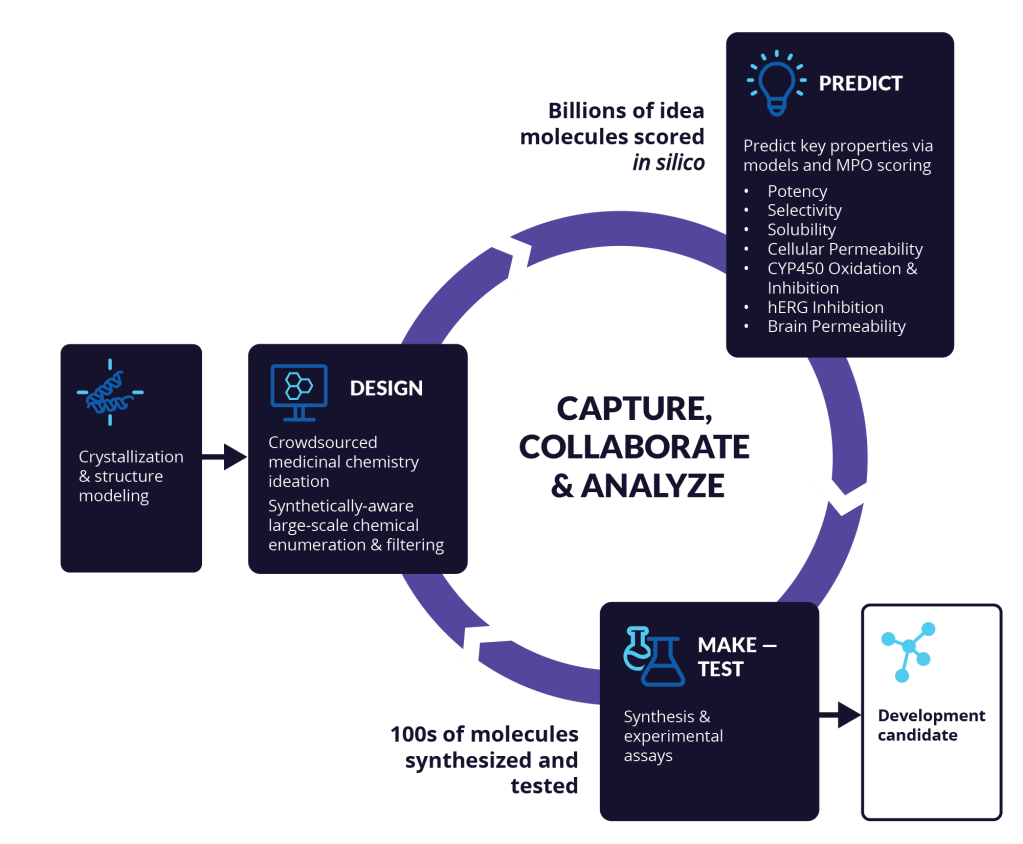
Explore and triage vast chemical space with high precision in silico tools
Identifying the best drug candidate — a novel molecule that optimizes key physicochemical properties while maintaining on-target potency and specificity — is the ultimate challenge of lead optimization programs.
Schrödinger’s platform for molecular design empowers project teams to deploy a ‘predict-first’ approach to lead optimization challenges, dramatically expanding the pool of molecules that can be explored through highly interactive, fully in silico design cycles. Teams can confidently spend time and energy exploring new, unknown, and often more complex designs while sending only the top performing molecules for synthesis.
Diverse solutions for chemical enumeration, property prediction, and team collaboration
Create and explore project-relevant chemical space to fast-track ligand design

Create and tailor your own chemical space using reaction or R-group based enumeration and advanced filtering capabilities
Combine accurate physics-based simulations with the power of machine learning to efficiently explore vast chemical space
Profile billions of virtual target-specific molecules with an intelligent, reaction-based enumeration, filtering and accurate FEP+ scoring workflow
Drive ligand design by leveraging the thermodynamics of water interactions in active sites
Discover new potency drivers by predicting the location and thermodynamic potential of hydration sites in the binding site
Visualize hydration sites for an easy and intuitive method of interpreting SAR
Design and collaborate in real-time with your colleagues — anytime, anywhere
Share, revise, and test design ideas with team members using a single cloud-native platform, LiveDesign
Capture decisions and hypotheses to improve collective SAR understanding and accelerate compound progression
Build rich dashboards to analyze whole project data or individual molecules and quickly identify promising design opportunities in key property space

Predict key properties to accelerate ligand optimization

Free energy-based computational assay (FEP+):
• Potency
• Selectivity
• Solubility
Other physics-based predictions:
• Membrane permeability
• hERG inhibition
• CYP inhibition / TDI
• CYP induction (DDI)
• Site of metabolism
• Brain exposure
Case Studies
Discover how Schrödinger technology is being used to solve real-world research challenges.
Hit to development candidate in 10 months: Rapid discovery of a novel, potent MALT1 inhibitor
Accelerating DMTA cycles with fast, push-button free energy calculations available to entire project teams
Morphic Therapeutic leverages digital chemistry strategy to design a novel small molecule inhibitor of α4β7 integrin
- Life Science
- Webinar
Design of a highly selective, allosteric, picomolar TYK2 inhibitor in clinical development
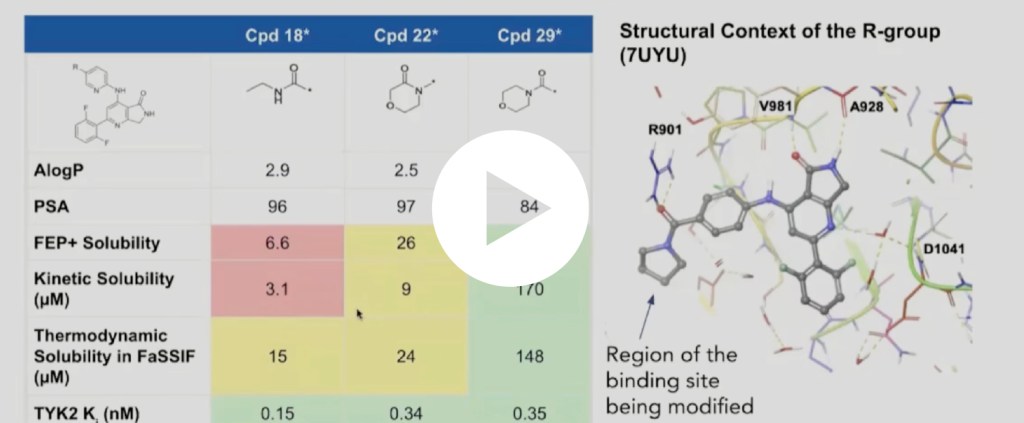
In this webinar, we highlight key moments from the discovery of this potentially best-in-class selective, allosteric, picomolar inhibitor of TYK2.
Watch webinar- Life Science
- Webinar
Impacting drug discovery programs with large-scale de novo design
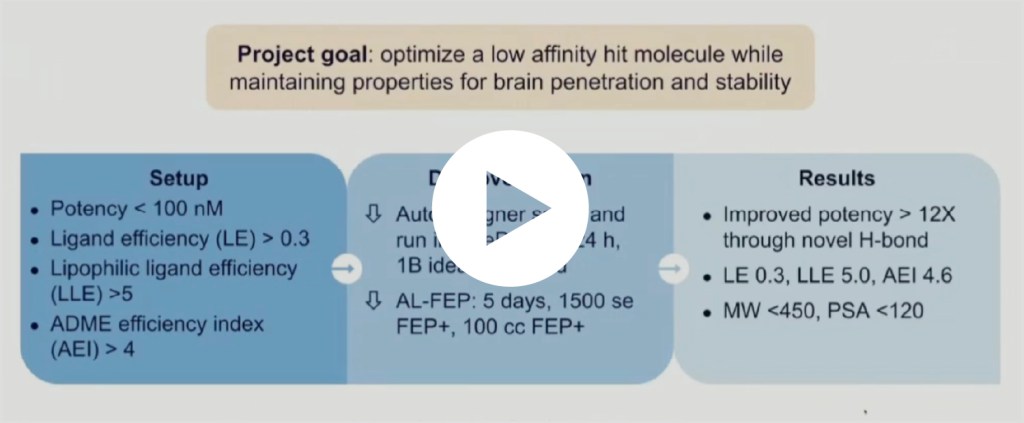
In this webinar, scientists from Schrödinger’s therapeutics group describe several recent case studies where de novo design technologies have allowed teams to overcome critical design challenges and accelerate programs.
Watch webinarKey Products
Learn more about the key computational technologies available to progress your research projects.
De Novo Design Workflow
Fully-integrated, cloud-based design system for ultra-large scale chemical space exploration and refinement
IFD-MD
Accurate ligand binding mode prediction for novel chemical matter, for on-targets and off-targets
WaterMap
State-of-the-art, structure-based method for assessing the energetics of water solvating ligand binding sites for ligand optimization
Publications
Browse the list of peer-reviewed publications using Schrödinger technology in related application areas.
Scaffold Hopping and Optimization of Small Molecule Soluble Adenyl Cyclase Inhibitors Led by Free Energy Perturbation
Sun, S. et al. J. Chem. Inf. Model. 2023, 63(9), 2828–2841
Discovery of a Novel Class of d-Amino Acid Oxidase Inhibitors Using the Schrödinger Computational Platform
Tang, H. et al. J. Med. Chem. 2022, 65(9), 6775–6802
AutoDesigner, a De Novo Design Algorithm for Rapidly Exploring Large Chemical Space for Lead Optimization: Application to the Design and Synthesis of d-Amino Acid Oxidase Inhibitors
Bos, P. H. et al. J. Chem. Inf. Model. 2022, 62(8), 1905–1915
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.