Dramatically improving hit rates with a modern virtual screening workflow
Scientists from Schrödinger’s Therapeutics Group leveraged a modern virtual screening workflow powered by ultra-large scale docking and absolute binding free energy calculations to achieve a double-digit hit rate for diverse protein targets.
Executive Summary
- Schrödinger developed a modern virtual screening workflow for small molecule ligands and fragments that enabled Schrödinger’s Therapeutics Group to repeatedly achieve unprecedented success in its hit discovery efforts
- The modern virtual screening workflow efficiently screens ultralarge libraries of up to several billion purchasable compounds with unrivaled accuracy, through machine learning enhanced docking and absolute binding free energy calculation technologies
- The workflow was successfully applied to a broad range of targets across multiple screening campaigns, for both whole ligands and fragments
- Schrödinger’s Therapeutics Group used the workflow to identify multiple experimentally confirmed hits with diverse chemotypes while dramatically narrowing down the number of compounds made or purchased and assayed in the lab — frequently achieving double-digit hit rates
Background
For years, hit discovery efforts using traditional virtual screening (VS) approaches have suffered from low hit rates, typically 1-2% in Schrödinger’s experience, which means that 100 compounds would have to be synthesized and assayed for 1-2 hits to be identified. These challenges have largely been attributed to two key factors:
First, traditional VS campaigns have been limited to libraries in the hundreds of thousands to a few million in size, providing limited coverage of chemical space. This is particularly critical for difficult-to-drug targets, where the random hit rate in the library is expected to be low, hence fewer hits are expected to be recovered with smaller libraries. In recent years, the emergence of ultra-large commercial chemical libraries such as Enamine REAL and research demonstrating the value of screening large libraries has further driven the need for technologies that can efficiently screen ultra-large chemical space.1
Second, traditional VS methods have been limited by the inaccuracy of the scoring methods utilized to rank order different ligands, such as GlideScore. Given a static view of the complex geometry and an approximate treatment of desolvation, such empirical scoring functions aren’t theoretically suited to quantitatively rank compounds by affinity. Thus, while docking is a powerful technology for early enrichment, ligand docking scores are not expected to and generally do not correlate with measured potency.
As a result of these limitations, most resources spent on virtual screens using traditional methods are often wasted. A more cost-effective and efficient approach to accurately screen ultra-large libraries is required to improve the success of virtual screens and ensure it is a viable path for hit discovery.
Design Approach
Over several years, Schrödinger’s Therapeutics Group has selected a number of challenging targets with the goal of identifying potent hit molecules. The group turned to a modern VS workflow, leveraging machine learning-guided Glide docking and highly accurate Absolute Binding FEP+ (ABFEP+) calculations, to screen and rescore ultra-large chemical libraries in a way that minimizes wet-lab costs and time while increasing the number and quality of hits available for hit-to-lead progression (Figure 1).
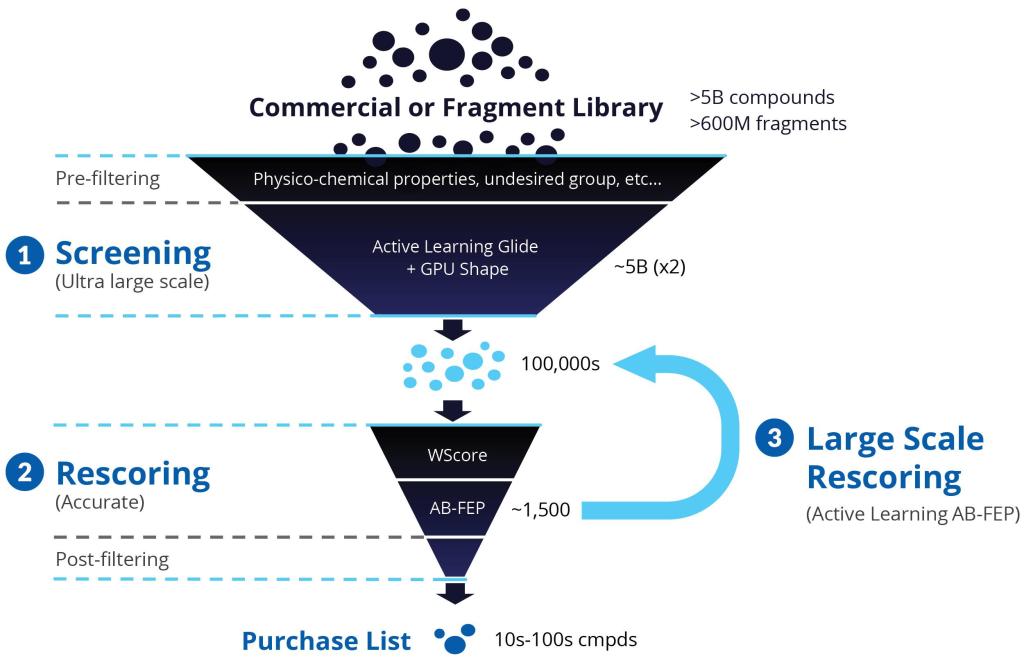
Step 1: Ultra-large scale screening for small molecule libraries
Starting with libraries on the order of several billion compounds (or libraries of up to 500 million for fragments), the team performed prefiltering based on physicochemical properties to eliminate any undesired groups. Next, they carried out a highthroughput virtual screen with Active Learning Glide (AL-Glide), in order to quickly identify the most promising compounds. Active learning is an effective supervised learning strategy that prioritizes training data for the next round of training based on a well-defined objective. AL-Glide combines machine learning (ML) with docking so that enrichment with docking can be applied to libraries of billions of compounds.2 By using this approach, only a fraction of the library is docked, reducing the computational cost significantly to a more reasonable level.
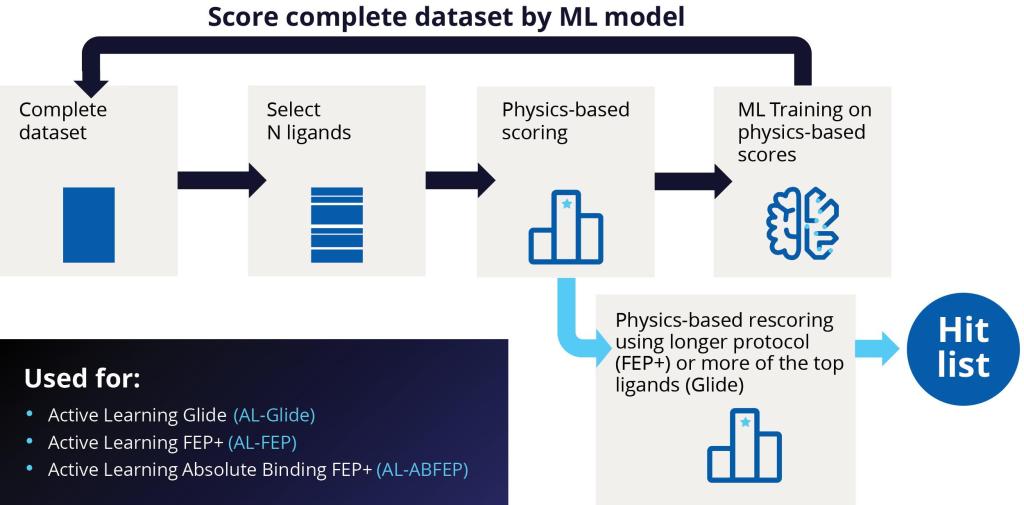
At the start of an active learning cycle, a manageable batch of compounds is selected from the complete data set of library compounds and docked. These selected compounds are then taken and added to the training set. The model is then trained on new information and continues to iterate this process as the machine learning model becomes a better and better proxy for the docking method (Figure 2).
This ML-guided docking model can evaluate compounds much more quickly than brute force docking. While the typical docking calculation with Glide might take an average of a few seconds per compound, the ML model can evaluate or make a prediction significantly faster, leading to a drastic increase in throughput. As a result, the ML-generated model is used to evaluate the entire library.
After completion of the AL-Glide screen, the team performed a full docking calculation using Glide on the best scored compounds, typically in the range of 10-100 million compounds.
Step 2: Rescoring
The most promising compounds based on Glide docking scores were then selected and subjected to a rescoring step using WScore, a sophisticated docking program that leverages explicit water information in the binding site to enrich active molecules over Glide alone, in large part due to improved pose prediction. WScore helps identify better compounds to pass to ABFEP+, the next scoring step, and to provide more reliable binding poses, reducing false positives in the process.3
Compounds with the best enrichment scores from docking are selected for rigorous rescoring with ABFEP+. ABFEP+ is a protocol in Schrödinger’s FEP+ technology that allows the accurate calculation of binding free energies between the bound and unbound states of the ligand/protein complex (Figure 3).4,5 ABFEP+ has proven to reliably correlate with experimentally measured binding affinities. Unlike relative binding FEP+, ABFEP+ does not require a similar, experimentally measured reference compound as a starting point. Because ABFEP+ can evaluate and accurately score diverse chemotypes, it is a linchpin technology to discover the most potent compounds in a virtual screen campaign.
Step 3: Large-Scale Rescoring
ABFEP+ is computationally expensive when compared to Relative Binding FEP+ (RB-FEP+), requiring multiple GPUs per ligand and approximately 4x more compute time. It is generally only practical to run thousands of ABFEP+ calculations on a hit discovery campaign. In order to realize the true enrichment benefit of ABFEP+, an active learning approach is utilized to score a much larger number of compounds.

* Compounds with molecular masses between 100 and 250 Da
Applying the modern virtual screening workflow to fragments
Experimental fragment screening has led to multiple FDA-approved drugs and clinical candidates. By adapting this modern workflow to fragment screening, Schrödinger’s Therapeutics Group has successfully scaled up screening to millions of fragments, as compared to 3k to 30k fragments screened by traditional HTS.
The in silico approach addresses a fundamental limitation of experimental fragment screening — the fragments need to be soluble enough to be assayed at high concentrations (100 μM to mM) against various targets. However, estimating the potency rigorously in silico is not limited by solubility, so the potency of the fragments can be assessed and subsequently pursued if they are predicted to be soluble enough given their estimated potency. The binding potency to the specific target is computed using active learning ABFEP+. Priority fragments are finally evaluated for solubility in silico at predicted potency using Solubility FEP+.6 In essence, the approach achieves scale by inverting the problem of potency and solubility, enabling the discovery of highly potent and ligand efficient fragments that would not exist in experimental fragment libraries.
To date, the team has completed a total of nine large fragment-based* virtual screens on multiple challenging targets, including one with a homology model. All nine screens yielded multiple potent ligand efficient hits, ranging from low nM to 30 μM in potency and double-digit hit rates.
Impact of Schrödinger’s modern VS workflow on hit rates across multiple projects and targets
Using these modern VS approaches, scientists at Schrödinger were able to demonstrate a drastic improvement in hit rates compared to traditional screens. As a result, several diverse hit compounds with high predicted binding affinity were identified, acquired, experimentally tested and confirmed as hits — resulting in an impressive doubledigit percentage hit rate (Figure 4).
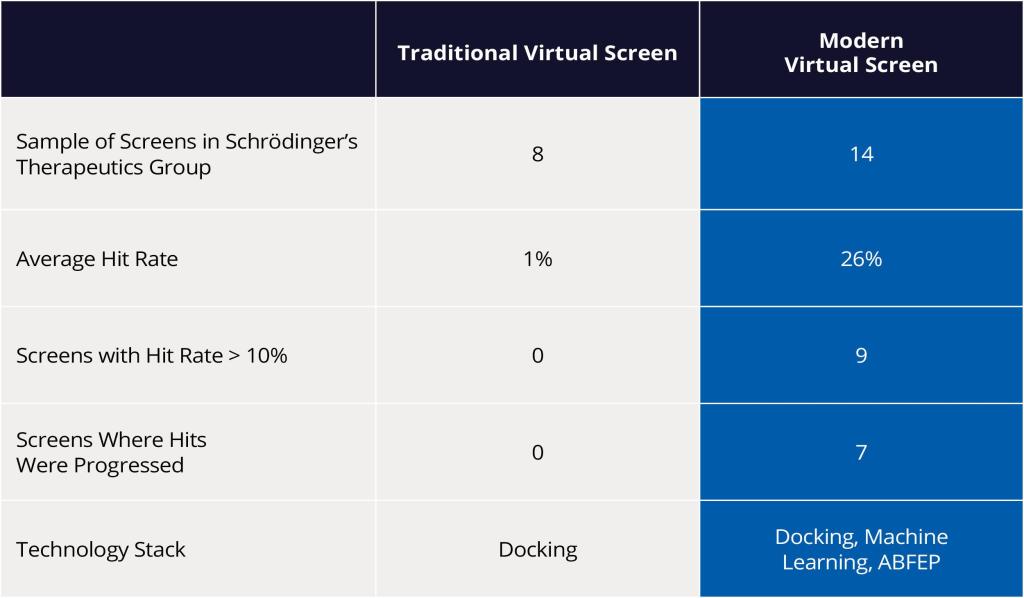
Conclusion
In silico hit identification has long relied on smaller scale libraries and lower accuracy methods to screen the chemical space, resulting in low hit rates and largely wasted wet lab resources.
By transitioning to a modern workflow that leverages rigorous physics-based methods, including absolute binding FEP+ combined with machine learning, Schrödinger’s Therapeutics Group has been able to successfully apply the workflow to a range of diverse targets across several projects and achieve a reproducible doubledigit hit rate. In the process, the team dramatically reduced the number of compounds synthesized and tested to reach the project’s lead candidate, reducing overall costs and project timelines.
This strategy empowers drug discovery teams by enabling them to explore the fast-growing ultra-large chemical libraries. It allows efficient navigation through the vast maze of chemical space, significantly improving the odds of identifying multiple hits with better properties and selectivity. Moreover, it accelerates the drug development process, leading to the faster discovery of higher-quality, novel drug candidates.
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting protein-ligand binding across broad chemical space at an accuracy matching experimental methods.
WScore
Advanced docking program that leverages explicit water information in the binding site to provide more accurate scoring of ligands.
Active Learning Glide
Powerful machine learning (ML) tool that trains ML to efficiently prioritize and select compounds for experimental evaluation or further screening.
References
-
Ultra-large library docking for discovering new chemotypes. Lyu J, et al.
Nature. 2019 Feb; 566(7743): 224–229.
-
Efficient exploration of chemical space with docking and deep learning. Yang Y, te al.
J. Chem. Theory Comput. 2021, 17, 11, 7106–7119.
-
WScore: A flexible and accurate treatment of explicit water molecules in ligand−receptor docking.
Murphy RB, et al. J. Med. Chem. 2016, 59, 4364−4384.
-
Enhancing hit discovery in virtual screening through accurate calculation of absolute protein-ligand binding free energies.
Chen W, et al. J. Chem. Inf. Model. 2023, 63, 10, 3171–3185.
-
Accurate calculation of the absolute free energy of binding for drug molecules.
Aldeghi M, et al. Chem. Sci. 2016;7:207–218.
-
Novel physics-based ensemble modeling approach that utilizes 3D molecular conformation and packing to access aqueous thermodynamic solubility: A case study of orally available bromodomain and extraterminal domain inhibitor lead optimization series.
Hong RS, et al. J. Chem. Inf. Model. 2021, 61, 3, 1412–1426.
Software and services to meet your organizational needs
Software Platform
Deploy digital materials discovery workflows with a comprehensive and user-friendly platform grounded in physics-based molecular modeling, machine learning, and team collaboration.
Research Services
Leverage Schrödinger’s expert computational scientists to assist at key stages in your materials discovery and development process.
Support & Training
Access expert support, educational materials, and training resources designed for both novice and experienced users.