Discovery of a novel, potent ACC inhibitor driven by computationally-guided design and assessment of water energetics in the binding site
Collaborative team of scientists from Nimbus Therapeutics and Schrödinger discover ND-630 for NASH and other liver diseases.
Leveraged virtual screening guided by hydration energetics to improve potency
Rapidly optimized ADME properties using structureguided methods
Resulted in the discovery of a development candidate in 16 months
ACC1 and ACC2
Collaborative program, small molecule
Type 2 diabetes, fatty liver disease, non-alcoholic steatohepatitis
Phase 2b clinical trial
“The discovery of ND-630 was one of the first demonstrations of the power of rigorous physics-based computational modeling to guide design and synthesis of a high-quality development candidate.”
Robert Abel
Chief Computational Officer
Schrödinger
Design challenge
As a key component of the fatty acid synthesis pathway (FaSyn), inhibition of acetyl-coenzyme A carboxylase (ACC) is a well validated strategy for the amelioration of metabolic disorders, such as type 2 diabetes, non-alcoholic steatohepatitis (NASH), and atherosclerotic vascular disease. ACC consists of two isoforms — ACC1 and ACC2, located in the cytoplasm and mitochondria, respectively. Additionally, ACC comprises two domains, the biotin carboxylase (BC) and carboxyltransferase (CT) domains, that carry out separate enzymatic functions.
Previous efforts have focused on inhibition of the CT domain, resulting in molecules with poor drug-like properties as well as selectivity issues. In comparison, targeting the BC domain represented an opportunity to develop a molecule with both improved properties and selectivity. Working together, scientific teams from Nimbus Therapeutics and Schrödinger leveraged a computationally-guided, structure-based approach to design novel drug candidates. The goal of this program was to identify potent and selective inhibitors of the BC domain of ACC1 and ACC2.
Identifying potent, selective inhibitors using a virtual screening workflow guided by hydration energetics
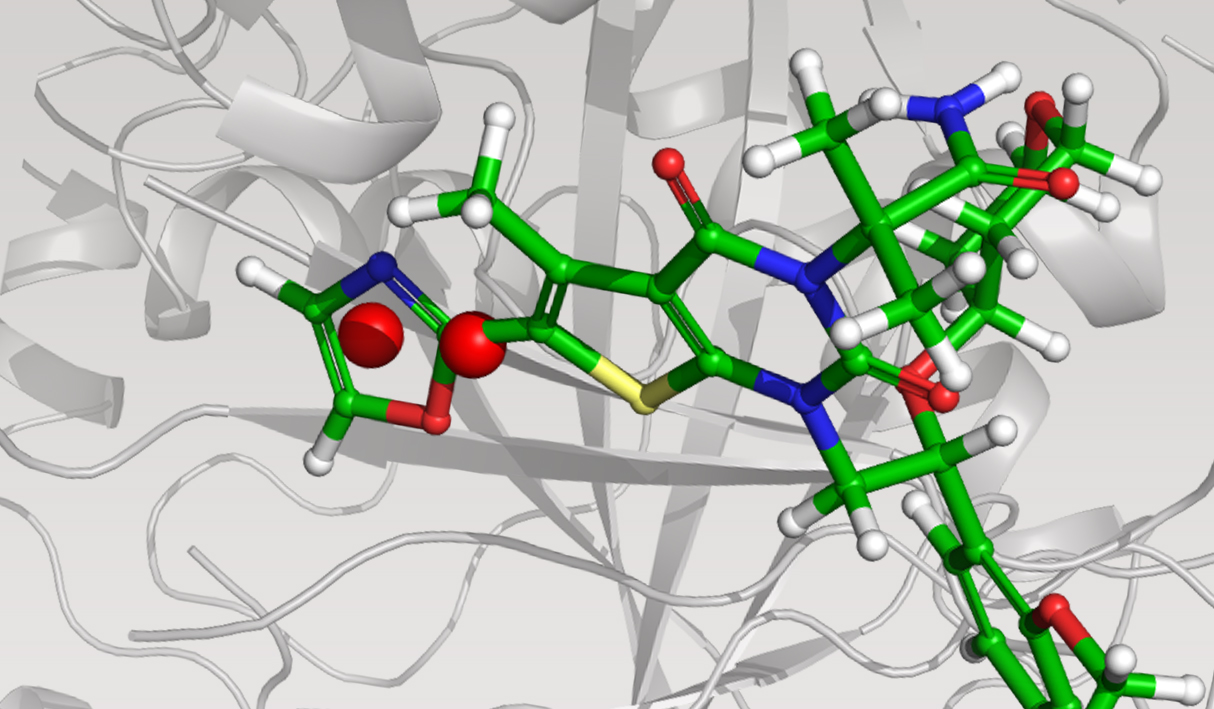
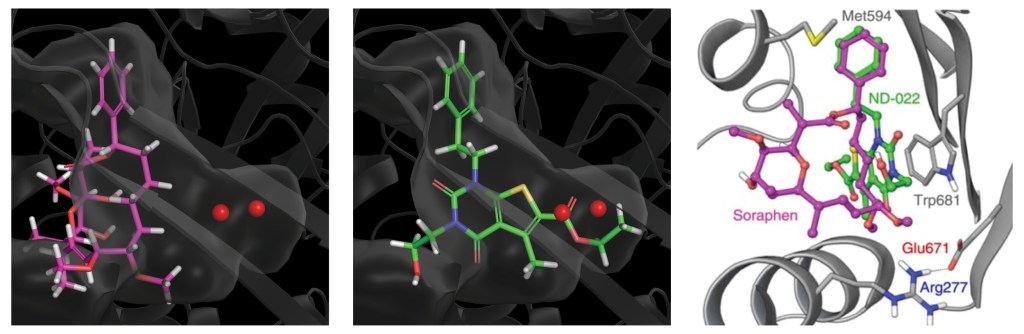
The team initiated the program with a two-pronged virtual screening workflow against the dimerization site of the BC domain. An available X-ray structure containing a bound ligand, Soraphen A, was used to guide both structure- and ligand-based screens for molecules that interfered with ligand binding and, consequently, enzymatic activity. Prior to conducting the screens, identification of high-energy hydration sites within the binding region was performed using WaterMap, a state-of-the-art, structure-based method for assessment of water energetics.1 Multiple high-energy hydration sites were identified by WaterMap; intriguingly, these sites were located in a deep, narrow pocket proximal to Val587 and Tyr683, and close to Soraphen A.
Using these hydration sites and other interactions as constraints, the team screened a commercially available library via docking with Glide, as well as pharmacophore screening with Phase (followed by docking). From this workflow, 250 chemically diverse molecules predicted to displace the high-energy hydration sites were evaluated in vitro. Ultimately, this strategy led to the identification of ND-022, a low micromolar dual inhibitor of ACC1 and ACC2 and the first molecule reported to mimic Soraphen A and inhibit enzyme activity via interaction within the ACC2 dimerization site (Figure 1).2
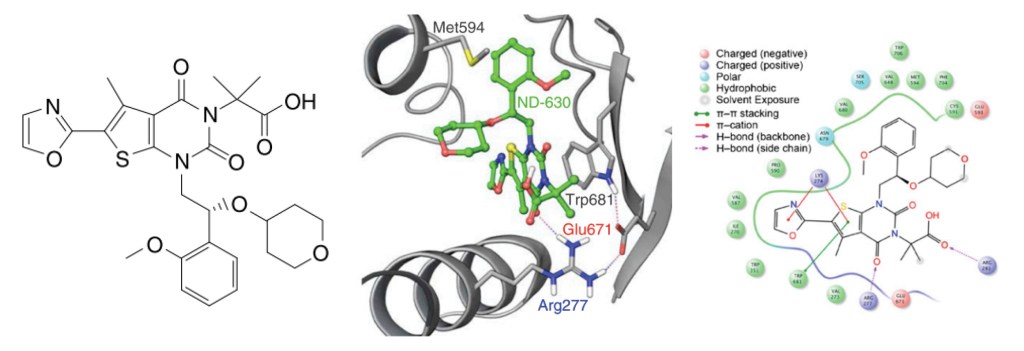
Rapid, structure-guided optimization of ADME properties results in clinical stage ACC inhibitor
Once a micromolar inhibitor was identified, the team focused their efforts on improving ACC2 inhibition, functional potency, and optimizing pharmacokinetics (PK). To achieve improved PK properties and potency, the team applied two complementary approaches – namely molecular mechanics and quantum mechanics. Guided by these simulations, four modifications were hypothesized to impact PK properties favorably while maintaining or improving potency. In only 16 months, this work culminated in the discovery of ND-630 (aka GS-0976, firsocostat), a potential first-in-class, potent, and selective inhibitor of the BC domain of ACC with favorable drug-like properties (Figure 2).2,3
In 2016, the program was acquired by Gilead Sciences.4 In 2017, Gilead presented positive data showing that a 20 mg orally administered dose of GS-0976/firsocostat, taken once daily for 12 weeks, led to statistically significant reductions in hepatic steatosis and the liver fibrosis marker TIMP-1, when compared to the placebo.5 Since then Gilead has prioritized development of GS-0976/firsocostat in combination with Novo Nordisk’s semaglutide, a GLP-1 receptor agonist.
In 2020, a five-arm trial evaluated combinations of Novo Nordisk’s semaglutide with Gilead’s investigational FXR agonist cilofexor and/or Gilead’s investigational ACC inhibitor firsocostat. The trial spanned 24 weeks and involved 108 patients with NASH. It successfully met its primary endpoint demonstrating that all regimens were well tolerated in patients with NASH and mild to moderate fibrosis, with the most common adverse events as gastrointestinal in nature.6
Furthermore, exploratory efficacy endpoints assessing biomarkers of liver health at 24 weeks in post-hoc analyses revealed statistically significant improvements in hepatic steatosis and liver injury in the combination arms versus semaglutide alone.6 Encouraged by these results, Gilead and Novo Nordisk are currently evaluating patients in a phase 2b trial for the combination.7 Although the clinical trial is ongoing, this case study highlights how drugs developed with Schrödinger technology have strong potential to impact the lives of patients with high unmet needs.
Enabling digital technologies to drive discovery programs
WaterMap
Structure-based method for assessing the energetics of water solvating ligand binding sites for ligand optimization.
References
-
Calculating water thermodynamics in the binding site of proteins – Applications of WaterMap to drug discovery.
Cappel et al. Curr. Top. Med. Chem. 2017,17(23), 2586-2598.
-
Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Harriman et al.
Proc. Natl. Acad. Sci. U S A. 2016 Mar 29, 113(13), E1796-805.
-
Accelerating drug discovery through tight integration of expert molecular design and predictive scoring.
Abel et al. Curr Opin Struct Biol. 2017, 43, 38-44.
-
Gilead Sciences announces acquisition of Nimbus Therapeutics’ acetyl-CoA carboxylase (ACC) program for NASH and other liver diseases.
Gilead. 2016.
-
Gilead announces Phase 2 results for GS-0976 in nonalcoholic steatohepatitis (NASH).
Gilead. 2017.
-
Gilead and Novo Nordisk present new data from proof-of-concept trial in NASH.
Gilead. 2020.
-
Gilead and Novo Nordisk expand NASH clinical collaboration.
Gilead. 2021.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.