Design of a novel, potent CDC7 inhibitor development candidate with high ligand efficiency and optimized properties
Use of a digital chemistry platform facilitates efficient multi-parameter optimization of selectivity, cell potency, and toxicity at scale.
compounds scored with rigorous physics-based modeling
total compounds synthesized
to discovery of development candidate
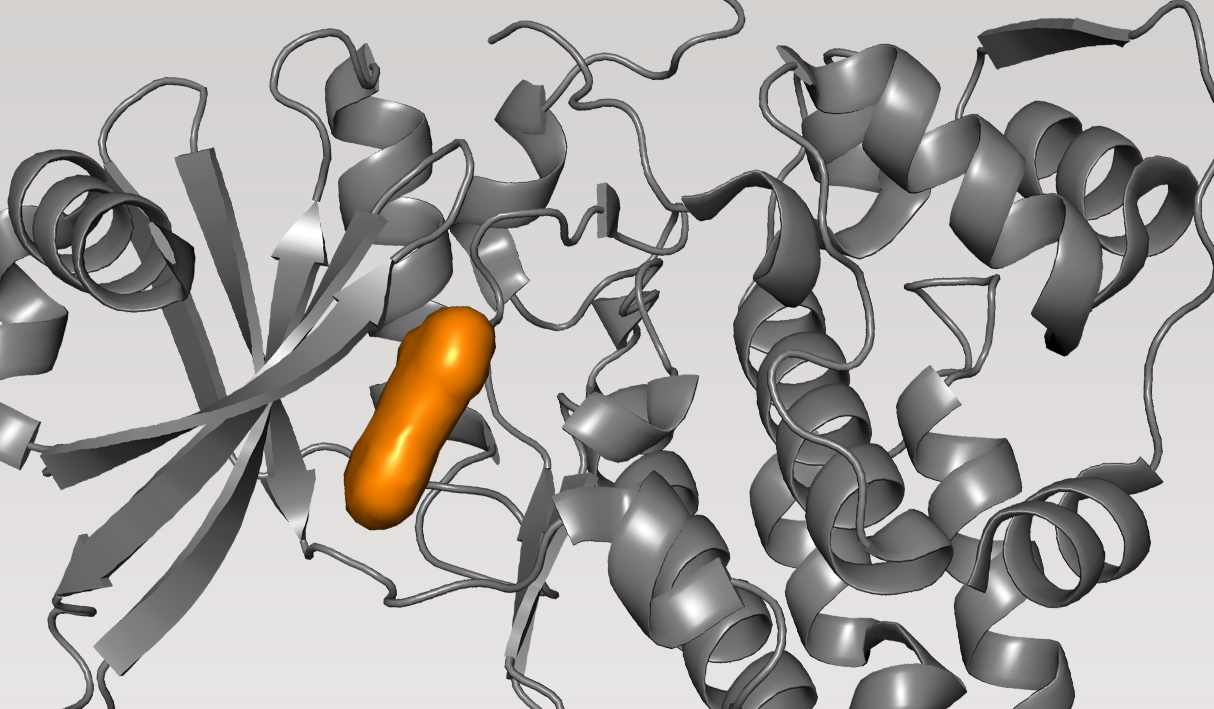
CDC7, kinase
Schrödinger proprietary program, small molecule
Hematological cancers and solid tumors
Phase 1 clinical trial
“Through large-scale chemical space exploration with our digital platform, the team was able to overcome several key program challenges, including a potential liability in the lead series. A quick pivot led to the discovery of SGR-2921, which is the most potent CDC7 inhibitor reported to date and possesses strong drug-like characteristics.”
Adam Levinson
Director, Medicinal Chemistry
Schrödinger Therapeutics Group
Design challenge
Cell division cycle 7-related protein kinase (CDC7) is a cell cycle kinase which plays a key role in replication stress response. Inhibition of CDC7 is a long pursued strategy for targeting hematological cancers and solid tumors. While drug discovery efforts focused on the development of small molecule inhibitors of CDC7 span more than 20 years, none have advanced beyond initial Phase I or II studies as most lacked the properties required for successful clinical development.
Given the very high affinity of CDC7 to its natural substrate, ATP, the primary challenge is the development of highly potent inhibitors — ideally picomolar, pM — to be substrate competitive while also balancing selectivity and other drug-like properties. Overall, the goal of the programwas to discover new chemotypes with best-in-class properties.
Better designs through crowdsourced ideation and physics-based modeling
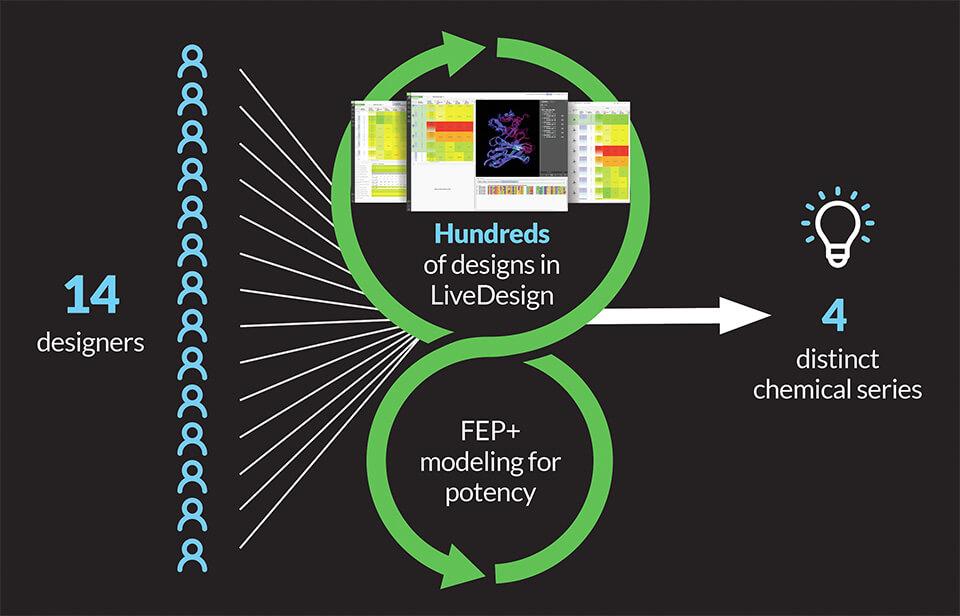
To rapidly iterate through high-quality ideas and analysis, the team crowdsourced ideation amongst 14 designers using LiveDesign, a modeling-enabled collaborative enterprise informatics platform for real-time design. Drawing on a wealth of existing structural and activity data, designs were evaluated through an iterative workflow using relative binding affinities via FEP+, free energy perturbation technology (Figure 1). The accuracy and utility of FEP+ as a computational assay for the prediction of relative binding energies of molecules has been validated extensively, generating predictions within 1.0 kcal/mol of experimental values on average.1 This armed the entire team with a true digital assay, allowing reliable predictions of potency and an unprecedented level of throughput.
Within four months of project start, the team identified four distinct, ligand-efficient chemical series with high affinity, ranging from low nanomolar to picomolar in biochemical assays. These chemotypes served as starting points for further optimization of potency and pharmacokinetic (PK) properties.
Overcoming selectivity and PK liabilities with next-generation modeling techniques
Given the structural similarity inherent within kinase families, achieving sufficient selectivity to mitigate offtarget effects is a core challenge of most kinase drug discovery programs. While the team successfully identified new, attractive chemical starting points, off-target mediated toxicity remained unresolved after the first round of design efforts. To address this selectivity liability, the team created a structural hypothesis for off-target binding using IFD-MD, Schrödinger’s state-of-the-art induced fit docking engine that generates highly accurate ligand binding poses in the absence of experimental structures. Poses resulting from IFD-MD were evaluated by FEP+ to ascertain potency as compared to CDC7. Ultimately, by utilizing this strategy the team identified a proof-of-concept compound with 10,000x selectivity for CDC7 against the off-target in question.
While selectivity was improved, the shift from biochemical potency to cell-based potency was large and variable; this could be attributed largely to suboptimal membrane permeability. A physics-based model for passive membrane permeability (Membrane Permeability) explained the discrepancy observed between experiments and enabled the team to prioritize designs with a consistently lower shift from biochemical to cell potency. Taking into consideration all factors required to satisfy the target product profile (TPP), the team deprioritized the lead series in favor of a new backup scaffold identified by FEP+ that exhibited a superior balance of properties including safety (Figure 2).
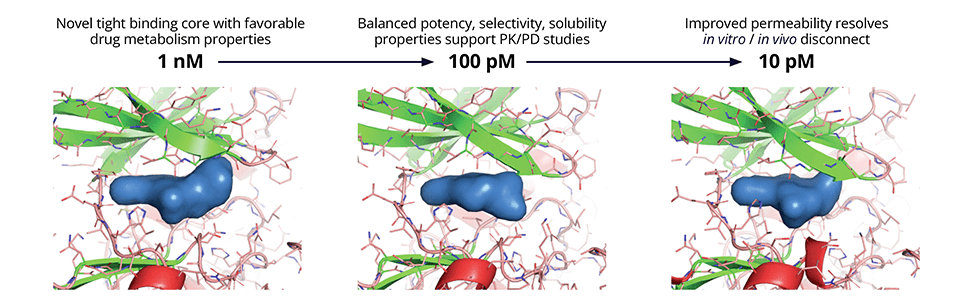
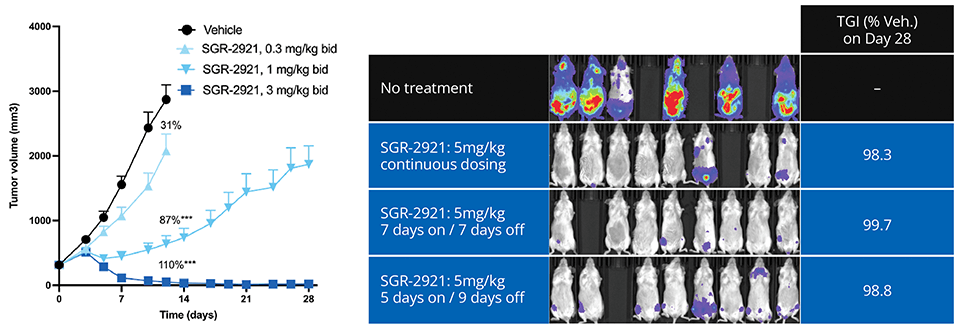
Ultimately, the team’s efforts culminated in the discovery of SGR-2921, with binding potency of ~10 pM in SPR with ligand efficiency > 0.6. SGR-2921 exhibits strong anti-tumor activity in vivo across multiple acute myeloid leukemia (AML) xenograft models, both as a monotherapy and in combination with standard of care agents (Figure 3). Moreover, SGR-2921 demonstrates potent anti-tumor activity in AML patient-derived samples that are resistant to standard of care agent.
Enabling digital technologies to drive discovery programs
FEP+
Digital assay for predicting protein-ligand binding across broad chemical space at an accuracy matching experimental methods.
IFD-MD
Accurate ligand binding mode prediction for novel chemical matter, for ontargets and off-targets.
Membrane Permeability
Physics-based, accurate predictions of passive membrane permeability.
LiveDesign
Collaborative enterprise informatics platform for centralizing access to virtual and wet lab project data and powerful computational predictions.
References
-
Advancing Drug Discovery Through Enhanced Free Energy Calculations.
Abel et al. Acc. Chem. Res. 2017, 50, 7, 1625–1632.
-
Inhibition of CDC7 with SGR-2921 in AML models results in enhanced DNA damage and anti-leukemic activity as monotherapy and in combination with standard of care agents.
Tsvetkov et al. ASH 2022.
-
Discovery of novel CDC7 inhibitors that disrupt cell cycle dynamics and show anti-proliferative effects in cancer cells.
Tsvetkov et al. AACR 2021.
-
Huang X, Mondal S, Ghanakota P, Boyles N, Frye L, Gerasyuto A, Greenwood JR, Tang H, Levinson AM. Cyclic Compounds and Methods of Using Same.
U.S. Patent No. WO/2021/113492. 2021.
-
Mondal S, Tang H,Huang X, Levinson AM, Frye L, Bhat S, Bos PH, Konst Z, Ghanakota P, Greenwood JR. Cyclic Compounds and Methods of Using Same.
U.S. Patent No. WO/2022/197898. 2022.
Software and services to meet your organizational needs
Industry-Leading Software Platform
Deploy digital drug discovery workflows using a comprehensive and user-friendly platform for molecular modeling, design, and collaboration.
Research Enablement Services
Leverage Schrödinger’s team of expert computational scientists to advance your projects through key stages in the drug discovery process.
Scientific and Technical Support
Access expert support, educational materials, and training resources designed for both novice and experienced users.